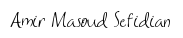
Understanding Factorized Top-K (FactorizedTopK) metric for recommendation systems with example
2022-04-30
Understanding Micro, Macro, and Weighted Averages for Scikit-Learn metrics in multi-class classification with example
2022-06-19In this post, I will provide the answers to the questions for the online ML interview book.
1) What are the basic assumptions to be made for linear regression?
Linear regression is a statistical method used to model the relationship between a dependent variable (often denoted as “y”) and one or more independent variables (often denoted as “x”). The basic assumptions to be made for linear regression are:
1. Linearity: The relationship between the dependent variable and independent variable(s) should be linear. This means that the rate of change in the dependent variable should be constant as the independent variable(s) change.
2. Independence: The observations should be independent of each other. This means that the value of one observation should not be influenced by the value of another observation.
3. Homoscedasticity: The variance of the errors should be constant across all levels of the independent variable(s). This means that the spread of the residuals (the differences between the predicted values and the actual values) should be consistent across all values of the independent variable(s).
4. Normality of Residuals: The errors (or residuals) should be normally distributed. This means that the distribution of the residuals should be symmetric around zero and follow a bell-shaped curve.
5. No multicollinearity: There should not be high correlation between independent variables. This means that each independent variable should be independent from the other independent variables used in the model.
Also, see this post:
https://www.analyticsvidhya.com/blog/2016/07/deeper-regression-analysis-assumptions-plots-solutions/
2) What happens if we don’t apply feature scaling to logistic regression?
Standardization isn’t required for logistic regression. The main goal of standardizing features is to help the convergence of the technique used for optimization. For example, if you use Newton-Raphson to maximize the likelihood, standardizing the features makes the convergence faster. Otherwise, you can run your logistic regression without any standardization treatment on the features.
If you use logistic regression with LASSO or ridge regression (as Weka Logistic class does) you should. As Hastie, Tibshirani, and Friedman point out (on page 82 of the pdf or on page 63 of the book):
If we don’t apply feature scaling to logistic regression, it can lead to some issues during the training process, and the model may not perform well. Here are a few reasons why:
1. Convergence: In logistic regression, the optimization algorithm tries to minimize the cost function. If the features are not scaled, some of them may have a much larger range of values than others. This can cause the optimization algorithm to take a long time to converge or not converge at all.
2. Outliers: If the features are not scaled, outliers in the data can have a large impact on the model. Since logistic regression uses the sigmoid function to map the output to a probability, the outliers can push the output towards 0 or 1, resulting in a less accurate model.
3. Interpretation: If the features are not scaled, it becomes harder to compare the coefficients of the logistic regression model. The magnitude of the coefficients will depend on the range of the corresponding features, and it will be difficult to interpret the relative importance of different features in the model.
In summary, feature scaling is an important step in logistic regression to ensure that the optimization algorithm converges efficiently, outliers do not have a disproportionate impact on the model, and the coefficients of the model can be interpreted more easily.
Generally, Not all machine learning algorithms require feature scaling. However, the following algorithms may benefit from feature scaling:
1. Gradient descent-based algorithms (e.g., linear regression, logistic regression, neural networks)
2. Distance-based algorithms (e.g., K-nearest neighbors, K-means clustering)
3. SVM (Support Vector Machine)
In general, feature scaling helps to improve the convergence rate and accuracy of these algorithms.
3) What are the algorithms you’d use when developing the prototype of a fraud detection model?
There are several machine learning algorithms that can be used for fraud detection. The choice of algorithm(s) depends on various factors such as the nature of the data, the size of the dataset, the complexity of the problem, and the level of interpretability required. Some of the commonly used algorithms for fraud detection include:
1. Logistic regression: This is a popular algorithm for binary classification tasks such as fraud detection. It is simple, interpretable, and can handle both categorical and continuous variables.
2. Decision trees: These are widely used for classification tasks and can handle both numerical and categorical data. Decision trees are easy to interpret, and can also capture interactions between variables.
3. Random forests: This is an ensemble algorithm that uses multiple decision trees to make predictions. Random forests are known to perform well on high-dimensional datasets and can capture complex non-linear relationships between variables.
4. Gradient boosting: This is another ensemble algorithm that combines multiple weak models to make a strong prediction. Gradient boosting is known for its ability to handle imbalanced datasets and its high predictive accuracy.
5. Support vector machines (SVMs): These are popular algorithms for binary classification tasks. SVMs can handle both linear and non-linear data and are known for their ability to handle high-dimensional datasets.
6. Neural networks: These are a class of algorithms inspired by the structure and function of the human brain. Neural networks are known for their ability to handle complex patterns in data and can be used for both regression and classification tasks.
It is worth noting that no single algorithm is best for all cases, and the choice of algorithm(s) should be based on the specific problem and dataset at hand.
4) Why do we use feature selection? What are some of the algorithms for feature selection? Pros and cons of each.
We use feature selection to improve the performance of a machine-learning model. By selecting the most relevant features, we can reduce the dimensionality of the data and improve the efficiency of the model. Additionally, feature selection can help prevent overfitting by removing irrelevant or redundant features, which can lead to a more accurate and generalizable model. Overall, feature selection can help improve the accuracy, speed, and interpretability of machine learning models.
Different types of feature selection techniques?
A. Here are some ways of selecting the best features out of all the features to increase the model performance, as the irrelevant features decrease the model performance of the machine learning or deep learning model.
– Filter Methods: Select features based on statistical measures such as correlation or chi-squared test.For example- Correlation-based Feature Selection, chi2 test, SelectKBest, and ANOVA F-value.
Pros: They are simple, computationally efficient, and can be used as a pre-processing step before applying more complex algorithms. Cons: They ignore the dependencies between features and may fail to identify important interactions between them.
– Wrapper Methods: These methods use a predictive model as a black-box to evaluate subsets of features and select the one that maximizes performance. For example- Recursive Feature Elimination, Backward Feature Elimination, Forward Feature Selection
Pros: They take into account the interactions between features and are more accurate than filter methods.
Cons: They are computationally expensive and may overfit the data.
– Embedded Methods: Select features by learning their importance during model training. For example- Lasso Regression, Ridge Regression, and Random Forest. These methods incorporate feature selection into the training of the model itself, by adding regularization terms that penalize the complexity of the model. Pros: They are computationally efficient and can improve the generalization performance of the model. Cons: They may require tuning of hyperparameters and may not be applicable to all types of models.
– Hybrid Methods: Combine the strengths of filter and wrapper methods. For Example- SelectFromModel
– Dimensionality Reduction Techniques: Reduce the dimensionality of the dataset and select the most important features. For Example- PCA, LDA, and ICA. Pros: They can improve the computational efficiency of the model and reduce overfitting. Cons: They may be harder to interpret and may require more training data.
5) How would you choose the value of k in k-means?
Choosing the optimal value of k in k-means clustering can be challenging and there is no definitive way to do it. Here are some common approaches:
1. Elbow Method: Plot the within-cluster sum of squares (WCSS) against the number of clusters (k). The elbow point in the graph is where the rate of decrease in WCSS slows down and forms an “elbow”. This point represents a good trade-off between reducing the sum of squares and increasing the number of clusters.
2. Silhouette Analysis: Compute the silhouette coefficient for each k. The silhouette coefficient measures how similar an object is to its own cluster compared to other clusters. Higher silhouette coefficients indicate better-defined clusters. Choose the k that maximizes the silhouette coefficient.
3. Domain Knowledge: If you have prior knowledge about the data and the problem you are trying to solve, it may help you to choose an appropriate value of k.
It is important to note that the chosen value of k may not always be the “correct” value, as there is often some degree of subjectivity involved. It is a good idea to try multiple values of k and compare the results to see which one makes the most sense for your particular problem.

Here’s an example of how to calculate the Silhouette score for a set of data points using the Euclidean distance metric:
Suppose we have a set of data points with the following coordinates in a 2-dimensional space:
(1, 2), (3, 4), (5, 6), (7, 8), (9, 10)
We perform k-means clustering on this data with k=2 (i.e., we aim to cluster the data points into two clusters).
After clustering, let’s say the first cluster has the following points:
(1, 2), (3, 4), (5, 6)
And the second cluster has the following points:
(7, 8), (9, 10)
We can calculate the Silhouette score for each data point as follows:
For the data point (1, 2) in the first cluster:
• Compute the average distance between (1, 2) and all other points in the first cluster (excluding itself): avg_dist_1 = (dist((1, 2), (3, 4)) + dist((1, 2), (5, 6))) / 2 = 2.828.
• Compute the average distance between (1, 2) and all points in the second cluster: avg_dist_2 = (dist((1, 2), (7, 8)) + dist((1, 2), (9, 10))) / 2 = 9.899.
• Compute the Silhouette score for (1, 2) as s = (avg_dist_2 – avg_dist_1) / max(avg_dist_1, avg_dist_2) = (9.899 – 2.828) / max(2.828, 9.899) = 0.657.
We can repeat this process for each data point in the two clusters to obtain the Silhouette score for each data point. Finally, we can compute the average Silhouette score for the entire clustering as the mean of the Silhouette scores for all data points.
6) If the labels are known, how would you evaluate the performance of your k-means clustering algorithm?
External validity indices are used to evaluate the performance of clustering algorithms based on some external criteria such as known class labels or expert domain knowledge. Some common external validity indices for clustering are:
External Validity indices for clustering:
1. Adjusted Rand Index (ARI)
2. Normalized Mutual Information (NMI)
3. Fowlkes-Mallows Index (FMI)
4. Jaccard Index
5. Precision, Recall, F1 Score
Internal Validity indices for clustering:
1. Silhouette Coefficient
2. Davies-Bouldin Index (DBI)
3. Calinski-Harabasz Index (CHI)
4. Dunn Index
5. Within-Cluster Sum of Squares (WSS)
If the labels are known, we can use external clustering evaluation metrics such as the Adjusted Rand index (ARI), normalized mutual information (NMI), or Fowlkes-Mallows index (FMI) to evaluate the performance of the k-means clustering algorithm.
Adjusted Rand index (ARI) measures the similarity between two clusterings, and the score ranges between -1 and 1. A score of 1 indicates perfect agreement between the two clusterings, while a score of 0 indicates random cluster assignments. A negative score indicates that the agreement is worse than random.
Normalized mutual information (NMI) measures the amount of information shared between the two clusterings, and the score ranges between 0 and 1. A score of 1 indicates that the two clusterings are identical, while a score of 0 indicates that the two clusterings are independent.
Fowlkes-Mallows index (FMI) is the geometric mean of precision and recall between two clusterings, and the score ranges between 0 and 1. A score of 1 indicates perfect agreement between the two clusterings, while a score of 0 indicates that the two clusterings are completely different.
To calculate these metrics, we can compare the labels assigned by k-means clustering to the true labels of the dataset and compute the score. The higher the score, the better the performance of the k-means clustering algorithm.
7) Given the following dataset, can you predict how K-means clustering works on it? Explain.
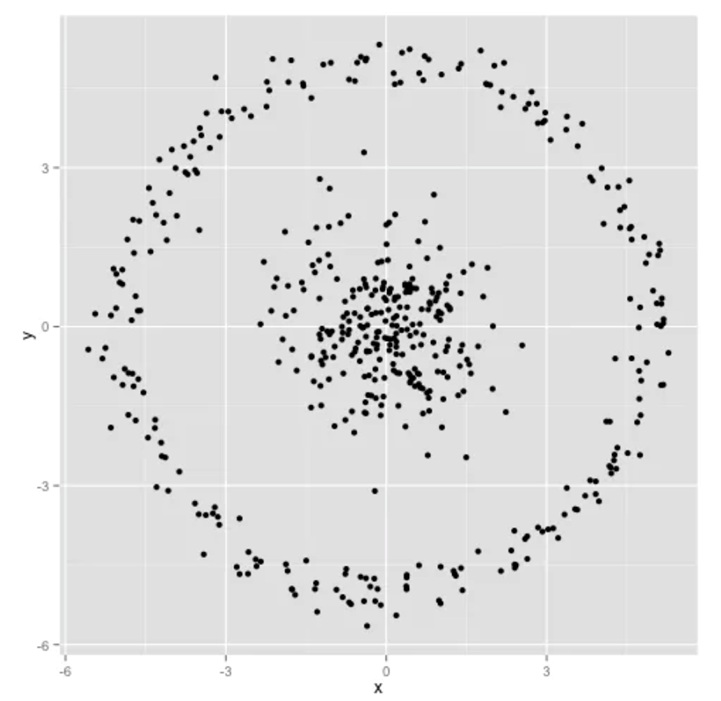
8) How would you choose the value of k in KNN?
There are various methods to choose the value of k in k-nearest neighbors, including:
1. Cross-validation: In this method, the dataset is divided into several folds, and the model is trained on each fold while validating it on the remaining folds. The performance of the model is evaluated for different values of k, and the optimal value of k is chosen based on the validation performance.
2. Grid search: In this method, the model is tested for different values of k, and the value that yields the best performance is chosen. This method can be computationally expensive, especially for larger datasets and high-dimensional feature spaces.
3. Rule of thumb: A commonly used rule of thumb is to set k to the square root of the number of samples in the dataset. However, this rule may not always lead to optimal performance and may need to be adjusted based on the problem at hand.
4. Domain knowledge: The value of k can also be chosen based on prior knowledge about the problem at hand. For example, if the problem involves detecting rare events, a smaller value of k may be more appropriate.
In practice, it is often necessary to try different values of k and evaluate their performance on a validation set to choose the optimal value of k for a particular problem.
9) What happens when you increase or decrease the value of k?
In the k-nearest neighbor (k-NN) classification, changing the value of k will have an impact on the model’s performance. Specifically, increasing or decreasing the value of k can lead to changes in the model’s bias and variance, which in turn can affect its accuracy and generalization ability.
When we increase the value of k, the decision boundary becomes smoother, and the model becomes less complex. As a result, the model’s variance decreases, but its bias increases. This means that the model becomes more likely to underfit the training data, which can lead to poor accuracy on both the training and test datasets.
On the other hand, when we decrease the value of k, the decision boundary becomes more complex, and the model becomes more flexible. As a result, the model’s variance increases, but its bias decreases. This means that the model becomes more likely to overfit the training data, which can lead to high accuracy on the training dataset but poor generalization performance on the test dataset.
Therefore, it is important to choose the appropriate value of k that balances the trade-off between bias and variance to achieve optimal performance on the test dataset. This can be done by using techniques such as cross-validation or grid search to find the best value of k for a particular problem.
10) How does the value of k impact the bias and variance?
In k-nearest neighbors (k-NN) classification, the value of k has an impact on the bias and variance of the model, which in turn affects its accuracy and generalization performance.
As we increase the value of k, the decision boundary becomes smoother and the model becomes less complex. This means that the model’s bias increases and its variance decreases. A higher value of k implies that the model considers a larger number of data points for each prediction, which leads to a more generalized model. However, a more generalized model may not capture the underlying patterns in the data, leading to underfitting. Therefore, as we increase the value of k, the model becomes more biased towards the class distribution in the dataset, which can result in lower accuracy.
On the other hand, as we decrease the value of k, the decision boundary becomes more complex, and the model becomes more flexible. This means that the model’s variance increases and its bias decreases. A lower value of k implies that the model considers fewer data points for each prediction, which leads to a more specific model. However, a more specific model may fit the training data too closely, leading to overfitting. Therefore, as we decrease the value of k, the model becomes less biased towards the class distribution in the dataset, which can result in higher accuracy.
smaller k => complex decision boundary / low bias / high variance
Larger k => smoother decision boundary / high bias / low variance
Thus, it is important to choose an appropriate value of k that balances the trade-off between bias and variance to achieve optimal performance on the test dataset. This can be done by using techniques such as cross-validation or grid search to find the best value of k for a particular problem.
11) Compare the k-means and GMM clustering algorithms. When would you choose one over another?
Hint: Here’s an example of how K-means and GMM algorithms perform on the artificial mouse dataset.
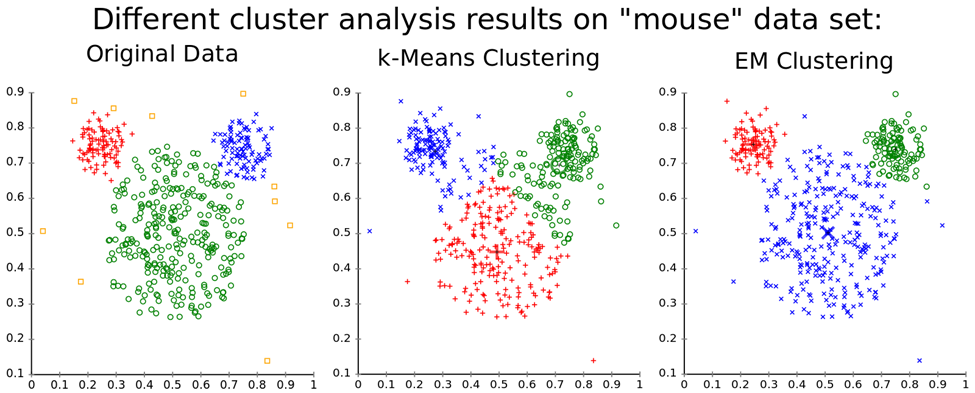
K-means and Gaussian Mixture Models (GMM) are both popular clustering algorithms used for unsupervised learning. Although they have similarities in terms of their clustering objectives, there are also several differences between them.
1. Algorithm Objective: K-means aims to partition a dataset into k clusters, where k is a predefined number. It works by iteratively assigning each data point to the nearest centroid and then updating the centroid based on the mean of the assigned points. K-means is a hard clustering algorithm, meaning that each data point is assigned to a single cluster, and the clusters are non-overlapping. It assumes that the clusters are spherical and have equal variances.
GMM, on the other hand, aims to model the underlying data distribution as a mixture of Gaussian distributions. It works by estimating the mean, covariance, and mixing coefficients of the Gaussian components using the Expectation-Maximization (EM) algorithm. GMM is a soft clustering algorithm, meaning that each data point is assigned a probability of belonging to each cluster, and the clusters can overlap. It can handle clusters of any shape and size and can capture complex data distributions.
2. Cluster Shape and Size: K-means assumes that the clusters are spherical and have equal variances, which means that it may not perform well on datasets with irregular shapes, varying cluster sizes, or noise. On the other hand, GMM can capture clusters of any shape and size and can handle noise.
3. Initialization: K-means requires initializations of the cluster centroids, which can affect the final clustering results. If the initialization is poor, the algorithm may converge to a suboptimal solution. GMM requires initialization of the mean, covariance, and mixing coefficients of the Gaussian components, which can also affect the final clustering results.
4. Convergence: K-means is guaranteed to converge to a local optimum, but it may not find the global optimum. The convergence rate of K-means is fast. GMM is more likely to converge to the global optimum, but the convergence rate is slower than K-means. GMM may also suffer from convergence issues if the data is high-dimensional or the number of Gaussian components is too large.
5. Performance and Scalability: K-means is computationally efficient and can handle large datasets with a large number of dimensions. However, it may not perform well on datasets with irregular shapes, varying cluster sizes, or noise. GMM is computationally more expensive than K-means and may require more computation resources and time. However, it can capture complex data distributions and can handle noise.
In summary, K-means is a simple and efficient algorithm suitable for datasets with well-separated spherical clusters, while GMM is a more complex and flexible algorithm suitable for datasets with complex distributions and overlapping clusters. The choice between the two depends on the problem at hand and the characteristics of the data.
When would you choose one over another?
The choice between k-means and GMM largely depends on the nature of the data and the specific problem at hand. Here are some general guidelines:
• If the data has well-defined clusters and the clusters are well-separated, k-means is likely to perform well. In contrast, GMM is better suited for datasets with overlapping clusters or where it is not clear which points belong to which cluster.
• If the data has a Gaussian distribution, GMM is likely to perform well as it can model the distribution of each cluster using a Gaussian function. In contrast, k-means makes the assumption that clusters are spherical and of equal size.
• If the number of clusters is known in advance, k-means is generally faster and more efficient. In contrast, GMM can estimate the number of clusters automatically using techniques such as the Bayesian information criterion (BIC).
• If the data is high-dimensional, k-means can suffer from the “curse of dimensionality,” making it difficult to find meaningful clusters. In contrast, GMM can handle high-dimensional data by modeling the covariance structure of the data.
Ultimately, the choice between k-means and GMM depends on the specific characteristics of the data and the goals of the analysis. It is often useful to try both algorithms and compare their results to determine which one performs better on a given dataset.
Steps of GMM:
Here are the steps of the GMM algorithm:
- Initialization: Choose the number of components (clusters) for the GMM and initialize their parameters. This includes selecting the initial values for the mean, covariance, and mixing coefficients of each Gaussian component.
- Expectation Step (E-step): Calculate the responsibilities or probabilities that each data point belongs to each component of the GMM. This is done by applying Bayes’ theorem and using the current parameter estimates.
- Maximization Step (M-step): Update the parameters of each Gaussian component based on the calculated responsibilities. This involves re-estimating the mean, covariance, and mixing coefficients using the weighted data points, where the weights are the responsibilities from the E-step.
- Evaluation: Compute the likelihood or log-likelihood of the data given the current GMM parameters. This provides a measure of how well the model fits the data.
- Convergence Check: Determine if the algorithm has converged by evaluating a convergence criterion. This can be done by comparing the log-likelihood between consecutive iterations or by checking the change in the parameters.
- Iteration: If the convergence criterion is not satisfied, go back to step 2 (E-step) and repeat the process of calculating responsibilities and updating the parameters until convergence is achieved.
- Finalization: Once the algorithm has converged, the estimated parameters of the GMM can be used for various purposes, such as clustering, density estimation, or generating new data samples from the learned distribution.
12) How determine the number of Gaussians for GMM?
Determining the number of Gaussian components (K) for a Gaussian Mixture Model (GMM) is an important consideration. Choosing the appropriate number of components is crucial to ensure that the model captures the underlying structure of the data without overfitting or underfitting. Here are a few methods commonly used to determine the number of Gaussians in a GMM:
- A priori knowledge or domain expertise: In some cases, you may have prior knowledge or domain expertise that can guide the choice of the number of components. For example, if you are working with a specific dataset or problem domain, you may have insights into the expected number of distinct clusters or modes in the data.
- Visualization and exploratory data analysis: It can be helpful to visualize the data and observe its distribution. Scatter plots, histograms, or density plots can provide insights into the number of distinct peaks or clusters present in the data. By visually inspecting the data, you may be able to make an initial estimation of the number of Gaussian components required.
- Information criteria: Information criteria, such as the Akaike Information Criterion (AIC) and the Bayesian Information Criterion (BIC), are statistical measures used to assess the goodness of fit of a model while considering the complexity of the model. These criteria balance the fit of the model with the number of parameters it uses. Lower values of AIC or BIC indicate a better fit. You can fit GMMs with different numbers of components and compare their corresponding AIC or BIC values. The number of components that minimizes the AIC or BIC can be chosen as the optimal number of Gaussians.
- Cross-validation: Cross-validation techniques can be employed to estimate the performance of the GMM for different numbers of components. The data is divided into training and validation sets, and GMMs with varying numbers of components are trained on the training set. The models’ performance is then evaluated on the validation set using a suitable metric, such as log-likelihood or mean squared error. The number of components that yields the best performance on the validation set can be selected.
- Elbow method: The elbow method is a heuristic approach that examines the rate of change of a metric, such as the sum of squared distances or the log-likelihood, as the number of components increases. A plot of the metric against the number of components is created, and the number of components at which the change in the metric starts to level off can be considered as a reasonable choice for the number of Gaussians.
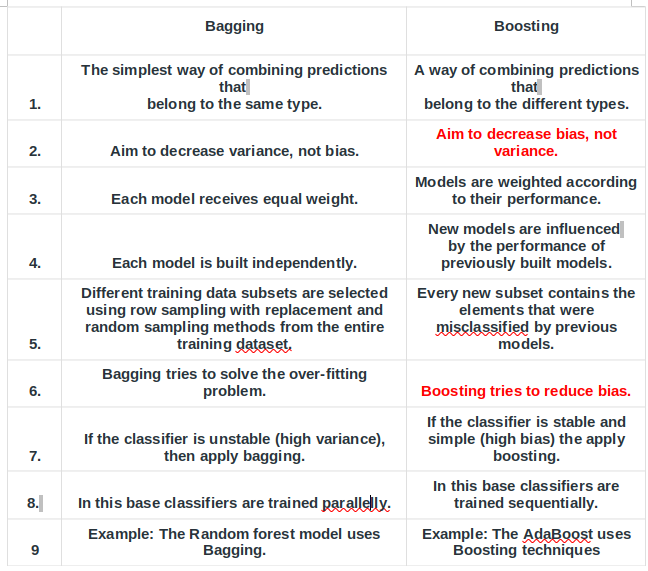
13) What are some of the fundamental differences between bagging and boosting algorithms? How are they used in deep learning?
Bagging and boosting are two popular ensembling methods with different approaches to combine multiple models to improve overall performance:
• Bagging (Bootstrap Aggregating) involves training multiple independent models on different subsets of the data by resampling the training set with replacement. The final prediction is then made by averaging the predictions of all models. Bagging helps reduce variance and prevent overfitting by increasing the diversity of the models.
• Boosting involves training multiple models sequentially where each subsequent model is trained to correct the errors made by the previous models. In other words, each model tries to improve the overall performance of the ensemble. Boosting can help reduce bias and improve accuracy by emphasizing the samples that are difficult to classify.
Some other differences between bagging and boosting include:
• Bagging is a parallel ensemble method, where each model is trained independently while boosting is a sequential ensemble method where each subsequent model is dependent on the previous ones.
• Bagging is more suitable for high variance models, while boosting is more suitable for high bias models.
• Bagging can improve the stability and robustness of the model by reducing the impact of outliers and noisy data, while boosting can improve the model’s accuracy by focusing on hard-to-predict samples.
Overall, bagging and boosting are both effective techniques for improving the performance of machine learning models, and the choice between them depends on the specific characteristics of the problem at hand and the type of model being used.
Bagging is best for data that has high variance, low bias, and low noise, as it can reduce overfitting and increase the model’s stability. Boosting is more suitable for data with low variance, high bias, and high noise, as it can reduce underfitting and increase accuracy.
Bagging and boosting techniques are not typically used in deep learning as they are more commonly applied to traditional machine learning models.
However, there are some variations of bagging and boosting that have been adapted for deep learning. For example:
• Dropout is a regularization technique that can be seen as a form of bagging. It involves randomly dropping out some neurons during training, effectively creating different sub-models that share some of the weights. Dropout helps reduce overfitting and improve the robustness of deep learning models.
• Stochastic Gradient Boosting is a variant of boosting that has been adapted for deep learning. It involves training multiple models sequentially, but instead of correcting the errors of the previous models, each subsequent model is trained on the residual errors of the previous ones. This approach can help improve the accuracy of deep learning models and has been used in some state-of-the-art models.
In general, deep learning models are already very complex and powerful, and therefore may not require the same level of ensembling techniques as traditional machine learning models. Instead, deep learning models often use regularization techniques such as dropout, weight decay, or early stopping to prevent overfitting and improve generalization performance.
14) Imagine we build a user-item collaborative filtering system to recommend to each user items similar to the items they’ve bought before. You can build either a user-item matrix or an item-item matrix. What are the pros and cons of each approach?
In a user-item collaborative filtering system, we can build either a user-item matrix or an item-item matrix to represent the relationships between users and items. Both approaches have their own advantages and disadvantages, which are summarized below:
User-item matrix:
• Pros:
◦ Intuitive representation of user-item relationships.
◦ Easy to understand and explain to others.
◦ Can handle new users easily by appending new rows to the matrix.
• Cons:
◦ Matrix can be very sparse if there are many items and few users, leading to scalability and storage issues.
◦ Difficult to compute similarities between users directly, as users may have rated different sets of items.
Item-item matrix:
• Pros:
◦ Sparse matrix problem is alleviated, as there are usually fewer items than users.
◦ Easier to compute similarities between items, as they have more common ratings by different users.
◦ Can capture more fine-grained item-item relationships, such as similar genres or features.
• Cons:
◦ Difficult to explain to others, as it may not be as intuitive as the user-item matrix.
◦ Cannot handle new items easily, as adding a new item would require recomputing the entire similarity matrix.
In summary, the choice between a user-item or item-item matrix depends on the specific use case and the trade-offs between scalability, interpretability, and the ability to handle new users and items.
15) How would you handle a new user who hasn’t made any purchases in the past?
Handling a new user in a user-item collaborative filtering system can be challenging because we don’t have any information about their preferences or behavior yet. One approach to handle this situation is to use a content-based filtering approach, where we make recommendations based on the characteristics of the items rather than the user’s past behavior.
Another approach is to use a hybrid recommendation system that combines collaborative filtering and content-based filtering. In this case, we can use the available information about the user to create a user profile based on demographic information or other attributes such as location or age. Then, we can use this profile to recommend items that are similar to items that other users with similar profiles have purchased in the past.
Alternatively, we can use a popular items approach, where we recommend the most popular items or the items with the highest ratings across all users. This approach is not personalized to the user but can be effective in the absence of user-specific information.
Overall, the best approach for handling a new user depends on the specific context and available data. A combination of different approaches may also be used to improve the recommendations.
16) Is feature scaling necessary for kernel methods?
Yes, feature scaling is usually necessary for kernel methods in order to improve their performance and accuracy.
Kernel methods, such as Support Vector Machines (SVM) and Kernel Ridge Regression (KRR), are sensitive to the scale of the input features. If the features are not scaled appropriately, it can lead to biased results or inaccurate predictions. For example, if one feature has a much larger scale than the others, it may dominate the kernel function and obscure the effects of other features.
Therefore, it is important to normalize or scale the features so that they have similar ranges and variances before applying kernel methods. This can be done using various methods such as standardization (mean=0, variance=1), min-max scaling (scaling features to a fixed range), or other normalization techniques.
17) What are kernel methods at all?
Kernel methods are a type of machine learning algorithm that operates on high-dimensional data by transforming the data into a higher-dimensional space using a kernel function. The transformed data can then be more easily separated or analyzed.
Kernel methods are used for a variety of tasks, including classification, regression, clustering, and dimensionality reduction. Some common examples of kernel methods include Support Vector Machines (SVM), Kernel Ridge Regression (KRR), and Gaussian Processes (GP).
The basic idea behind kernel methods is to find a function that maps the input data into a higher-dimensional space where it is easier to separate or analyze. This is done by defining a kernel function that measures the similarity between pairs of data points in the input space. The kernel function is then used to compute the dot product of the transformed data points in the higher-dimensional space, without actually computing the transformation explicitly.
Kernel methods have several advantages over other machine learning algorithms. They can handle high-dimensional and non-linear data, and they can generalize well to unseen data. They are also relatively easy to implement and can be applied to a wide range of problems.
18) How is the Naive Bayes classifier naive?
The Naive Bayes classifier is considered “naive” because it makes a simplifying assumption that the features or attributes used to describe an instance are conditionally independent, given the class label. This means that the presence or absence of a particular feature is independent of the presence or absence of any other feature, given the class label.
In other words, it assumes that the features are independent of each other, which is often not the case in the real world. However, despite this naive assumption, Naive Bayes has been shown to perform well in many practical applications, particularly when the number of features is large. This is because it is computationally efficient and can handle high-dimensional data well.
The Naive Bayes classifier is based on Bayes’ theorem, which is a fundamental rule in probability theory. Bayes’ theorem states that:
P(A|B) = P(B|A) * P(A) / P(B)
where P(A|B) is the probability of A given B, P(B|A) is the probability of B given A, P(A) is the prior probability of A, and P(B) is the prior probability of B.
In the case of the Naive Bayes classifier, A represents the class label, and B represents the features or attributes of the instance. The classifier uses Bayes’ theorem to calculate the probability of the class label given the features:
P(y|x1, x2, …, xn) = P(x1, x2, …, xn|y) * P(y) / P(x1, x2, …, xn)
where y is the class label, x1, x2, …, xn are the features or attributes, and P(y|x1, x2, …, xn) is the posterior probability of the class label given the features.
The key assumption in the Naive Bayes classifier is that the features are conditionally independent given the class label. This means that we can express the joint probability of the features given the class label as the product of the individual probabilities of each feature given the class label:
P(x1, x2, …, xn|y) = P(x1|y) * P(x2|y) * … * P(xn|y)
This assumption simplifies the calculation of the posterior probability and makes the classifier computationally efficient.
To classify a new instance, the Naive Bayes classifier calculates the posterior probability of each class label given the features and selects the class label with the highest probability.
19) Let’s try to construct a Naive Bayes classifier to classify whether a tweet has a positive or negative sentiment. We have four training samples: According to your classifier, what’s the sentiment of the sentence The hamster is upset with the puppy?
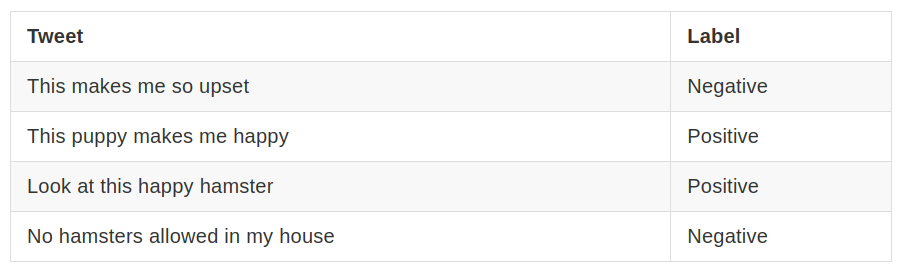
Let’s try to construct a Naive Bayes classifier to classify whether a tweet has a positive or negative sentiment. We have four training samples: Tweet Label This makes me so upset Negative This puppy makes me happy Positive Look at this happy hamster Positive No hamsters allowed in my house Negative 13. According to your classifier, what’s the sentiment of the sentence The hamster is upset with the puppy?
ChatGPT
To classify the sentence “The hamster is upset with the puppy”, we need to first extract its features. In this case, we can use the presence or absence of certain words as features, such as “hamster”, “puppy”, “upset”, and “happy”.
Assuming we use these four words as our features, we can calculate the likelihood of each feature given a positive or negative sentiment label based on the training data. For example:
• P(“hamster” | Positive) = 1/2
• P(“hamster” | Negative) = 0
• P(“puppy” | Positive) = 1/2
• P(“puppy” | Negative) = 1/2
• P(“upset” | Positive) = 0
• P(“upset” | Negative) = 1/2
• P(“happy” | Positive) = 1
• P(“happy” | Negative) = 0
Next, we need to calculate the prior probabilities of each sentiment label based on the training data:
• P(Positive) = 2/4 = 0.5
• P(Negative) = 2/4 = 0.5
Using Bayes’ theorem, we can calculate the posterior probabilities of each label given the features of the sentence:
• P(Positive | “hamster” “puppy” “upset”) ∝ P(“hamster” | Positive) * P(“puppy” | Positive) * P(“upset” | Positive) * P(Positive) = 0
• P(Negative | “hamster” “puppy” “upset”) ∝ P(“hamster” | Negative) * P(“puppy” | Negative) * P(“upset” | Negative) * P(Negative) = 0
Therefore, the Naive Bayes classifier would classify the sentence “The hamster is upset with the puppy” as unclassified or possibly as a neutral sentiment since both posterior probabilities are 0.
Laplacian Version
In Naive Bayes classification, Laplacian smoothing (also called additive smoothing) is a technique used to handle the problem of zero-frequency.
When estimating the probability of a feature value given a class, it is possible that some feature values may not occur in the training set for a particular class. This can lead to zero probabilities, making the classifier unable to make predictions for that feature. Laplacian smoothing adds a small value (usually 1) to the count of each feature value in each class, effectively assuming that each feature value occurs at least once in each class. This avoids zero probabilities and allows the classifier to make predictions for all features.
The formula for Laplacian smoothing is as follows:
P(x_i|y) = (count(x_i, y) + alpha) / (count(y) + alpha * n)
where P(x_i|y) is the probability of feature x_i given class y, count(x_i, y) is the count of feature x_i in class y, count(y) is the total count of class y, alpha is the smoothing parameter (usually set to 1), and n is the number of possible feature values.
To use Laplacian smoothing in Naive Bayes, we add a small constant value (usually 1) to each count in the probability calculation.
Let’s assume we use Laplacian smoothing with k=1. We need to calculate the probabilities of each word given the positive or negative class:
Positive:
• P(hamster | positive) = (1+1) / (3+14) = 2/7 • P(puppy | positive) = (1+1) / (3+14) = 2/7
• P(happy | positive) = (2+1) / (3+14) = 3/7 Negative: • P(this | negative) = (1+1) / (2+14) = 2/7
• P(makes | negative) = (1+1) / (2+14) = 2/7 • P(me | negative) = (1+1) / (2+14) = 2/7
• P(so | negative) = (1+1) / (2+14) = 2/7 • P(upset | negative) = (1+1) / (2+14) = 2/7
• P(no | negative) = (1+1) / (2+14) = 2/7 • P(allowed | negative) = (1+1) / (2+14) = 2/7
• P(in | negative) = (1+1) / (2+14) = 2/7 • P(my | negative) = (1+1) / (2+14) = 2/7
• P(house | negative) = (1+1) / (2+1*4) = 2/7
Now, we can use these probabilities to calculate the sentiment of the sentence “The hamster is upset with the puppy” using the Naive Bayes formula:
Positive probability:
• P(positive | hamster, upset, puppy) = P(hamster | positive) * P(upset | positive) * P(puppy | positive) * P(positive) = (2/7) * (1/3) * (2/7) * (3/4) = 0.0204
Negative probability:
• P(negative | hamster, upset, puppy) = P(hamster | negative) * P(upset | negative) * P(puppy | negative) * P(negative) = (1/7) * (2/7) * (1/7) * (1/4) = 0.0007
Since the positive probability is higher, the Naive Bayes classifier would predict that the sentiment of the sentence “The hamster is upset with the puppy” is positive.
20) Two popular algorithms for winning Kaggle solutions are Light GBM and XGBoost. They are both gradient boosting algorithms. What is gradient boosting? What problems is gradient boosting good for?
What is gradient boosting?
Gradient Boosting is a popular machine learning technique used for both regression and classification tasks. It involves the creation of a predictive model in a step-wise fashion by iteratively adding weak models to the ensemble.
In gradient boosting, each new weak model is trained on the residuals (the difference between the actual and predicted values) of the previous model. This approach allows the new models to focus on the patterns in the data that were missed by the previous models, improving the overall accuracy of the final ensemble.
What problems is gradient boosting good for?
Gradient Boosting is particularly effective in solving problems with large and complex datasets. It is commonly used for problems with high-dimensional feature spaces, non-linear interactions between features, and noisy or missing data. Gradient Boosting has been applied to a wide range of tasks, including image and speech recognition, natural language processing, and financial forecasting.
https://towardsdatascience.com/lightgbm-vs-xgboost-which-algorithm-win-the-race-1ff7dd4917d
Forms of Boosting
Boosting can take several forms, including:
- Adaptive Boosting (Adaboost)
Adaboost aims at combining several weak learners to form a single strong learner. Adaboost concentrates on weak learners, which are often decision trees with only one split and are commonly referred to as decision stumps. The first decision stump in Adaboost contains observations that are weighted equally.
Previous errors are corrected, and any observations that were classified incorrectly are assigned more weight than other observations that had no error in classification. Algorithms from Adaboost are popularly used in regression and classification procedures. An error noticed in previous models is adjusted with weighting until an accurate predictor is made. - Gradient Boosting
Gradient boosting, just like any other ensemble machine learning procedure, sequentially adds predictors to the ensemble and follows the sequence in correcting preceding predictors to arrive at an accurate predictor at the end of the procedure. Adaboost corrects its previous errors by tuning the weights for every incorrect observation in every iteration. Still, gradient boosting aims at fitting a new predictor in the residual errors committed by the preceding predictor.
Gradient boosting utilizes the gradient descent to pinpoint the challenges in the learners’ predictions used previously. The previous error is highlighted, and by combining one weak learner to the next learner, the error is reduced significantly over time. - XGBoost (Extreme Gradient Boosting)
XGBoostimg implements decision trees with boosted gradient, enhanced performance, and speed. The implementation of gradient boosted machines is relatively slow due to the model training that must follow a sequence. They, therefore, lack scalability due to their slowness.
XGBoost is reliant on the performance of a model and computational speed. It provides various benefits, such as parallelization, distributed computing, cache optimization, and out-of-core computing.
XGBoost provides parallelization in tree building through the use of the CPU cores during training. It also distributes computing when it is training large models using machine clusters. Out-of-core computing is utilized for larger data sets that can’t fit in the conventional memory size. Cache optimization is also utilized for algorithms and data structures to optimize the use of available hardware.
Pros and Cons of Boosting
As an ensemble model, boosting comes with an easy-to-read and interpret algorithm, making its prediction interpretations easy to handle. The prediction capability is efficient through the use of its clone methods, such as bagging or random forest and decision trees. Boosting is a resilient method that curbs over-fitting easily.
One disadvantage of boosting is that it is sensitive to outliers since every classifier is obliged to fix the errors in the predecessors. Thus, the method is too dependent on outliers. Another disadvantage is that the method is almost impossible to scale up. This is because every estimator bases its correctness on the previous predictors, thus making the procedure difficult to streamline.
21) What’s linear separation? Why is it desirable when we use SVM?
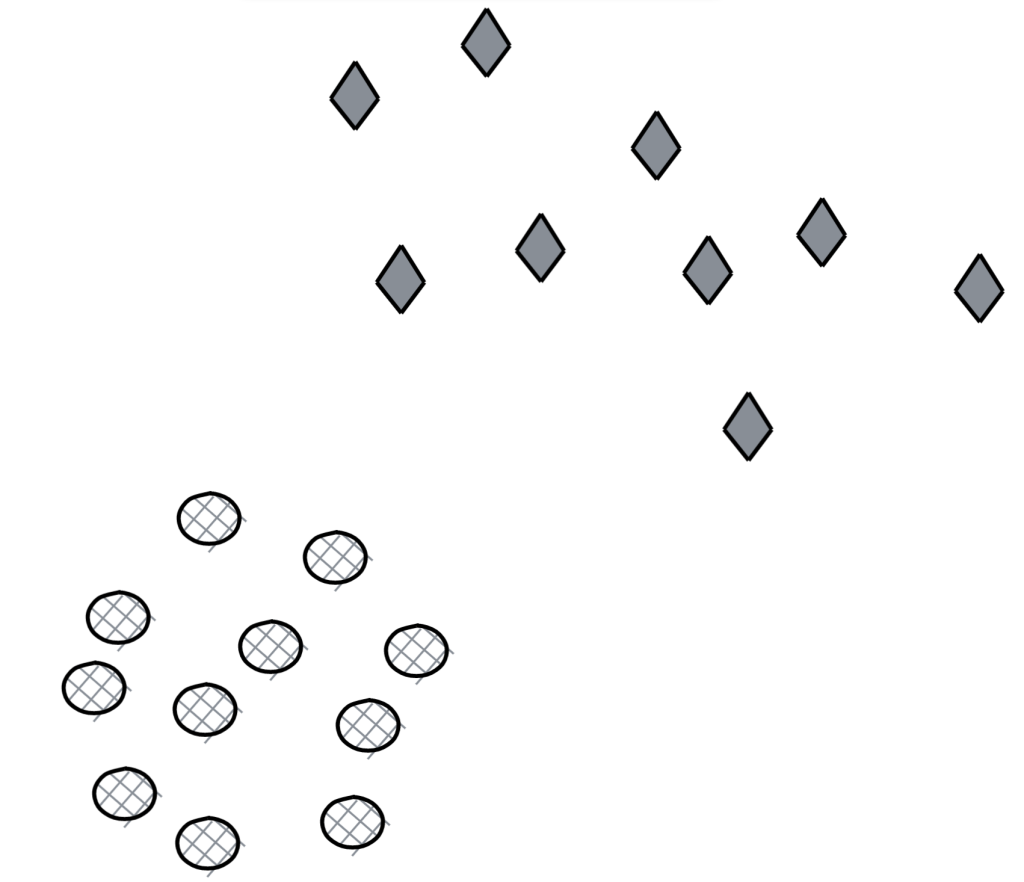
Linear separation is a property of a dataset where the two classes can be separated by a straight line or a hyperplane in a higher dimensional space. Linear separation is desirable when we use SVM because the SVM algorithm is designed to find the hyperplane that separates the two classes with the largest margin. When the data is linearly separable, it is easier to find a hyperplane with a large margin, and this hyperplane can generalize well to new data. In contrast, if the data is not linearly separable, then finding the hyperplane with the largest margin is not possible, and some trade-off must be made between the size of the margin and the number of misclassified examples. Therefore, linearly separable data is desirable for SVM as it leads to better classification performance
- [M] How well would vanilla SVM work on this dataset?
- [M] How well would vanilla SVM work on this dataset?
- [M] How well would vanilla SVM work on this dataset?
Margin:
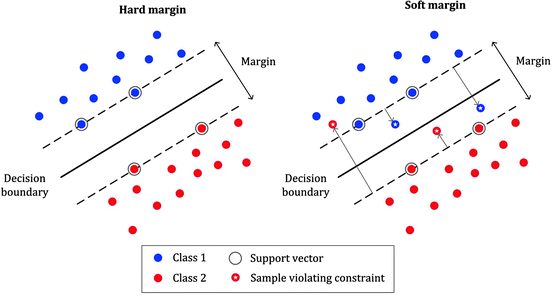
In Support Vector Machines (SVM), the margin refers to the separation between the decision boundary (hyperplane) and the closest data points from each class. The SVM algorithm aims to find an optimal hyperplane that maximizes this margin.
To understand the margin concept in SVM, let’s consider a binary classification problem where we have two classes, class A and class B. The goal is to find a hyperplane that best separates the two classes in the feature space.
The margin is defined as the perpendicular distance between the decision boundary and the closest data points from each class, also known as support vectors. These support vectors are the data points that are closest to the decision boundary and play a crucial role in defining the hyperplane.
The margin is important because it represents the generalization capability of the SVM model. A larger margin implies better separation between classes and indicates a more robust decision boundary. The intuition behind maximizing the margin is to improve the model’s ability to classify unseen data accurately.
The SVM algorithm seeks to find the hyperplane that maximizes the margin while still correctly classifying the training data. This process involves solving an optimization problem to find the optimal hyperplane parameters. The hyperplane is defined by a weight vector and a bias term, which are determined by the SVM algorithm during training.
In summary, the margin in SVM is the separation between the decision boundary and the closest data points from each class. Maximizing this margin is a key objective of the SVM algorithm to achieve better classification performance and improve the model’s generalization ability.
SVM Loss:
